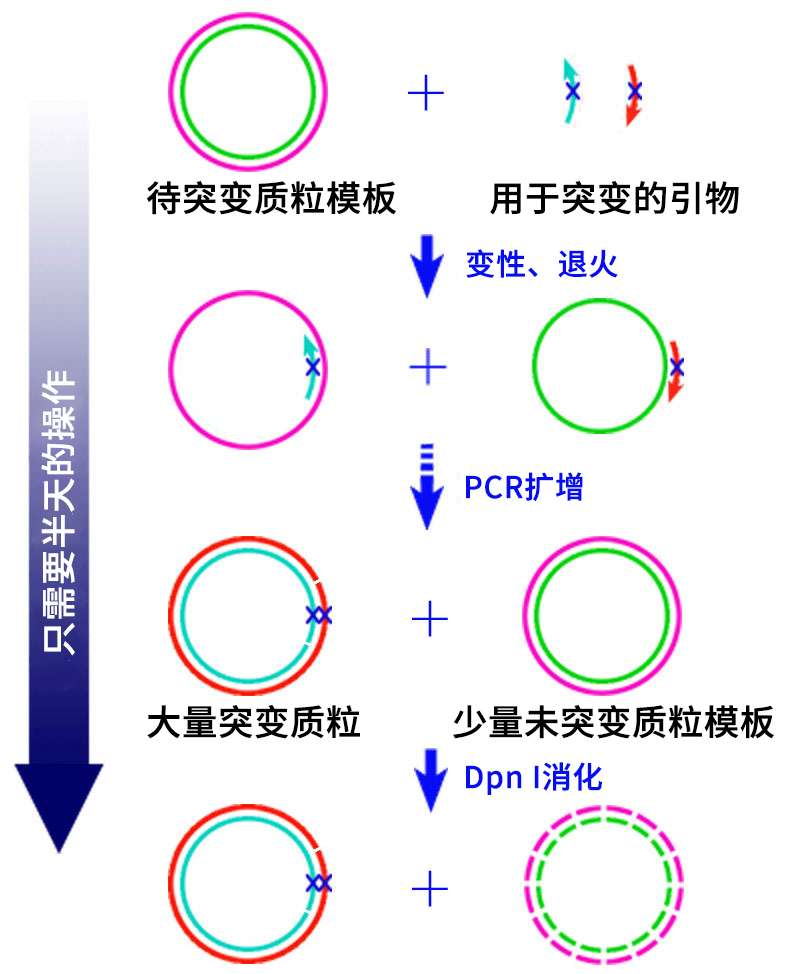
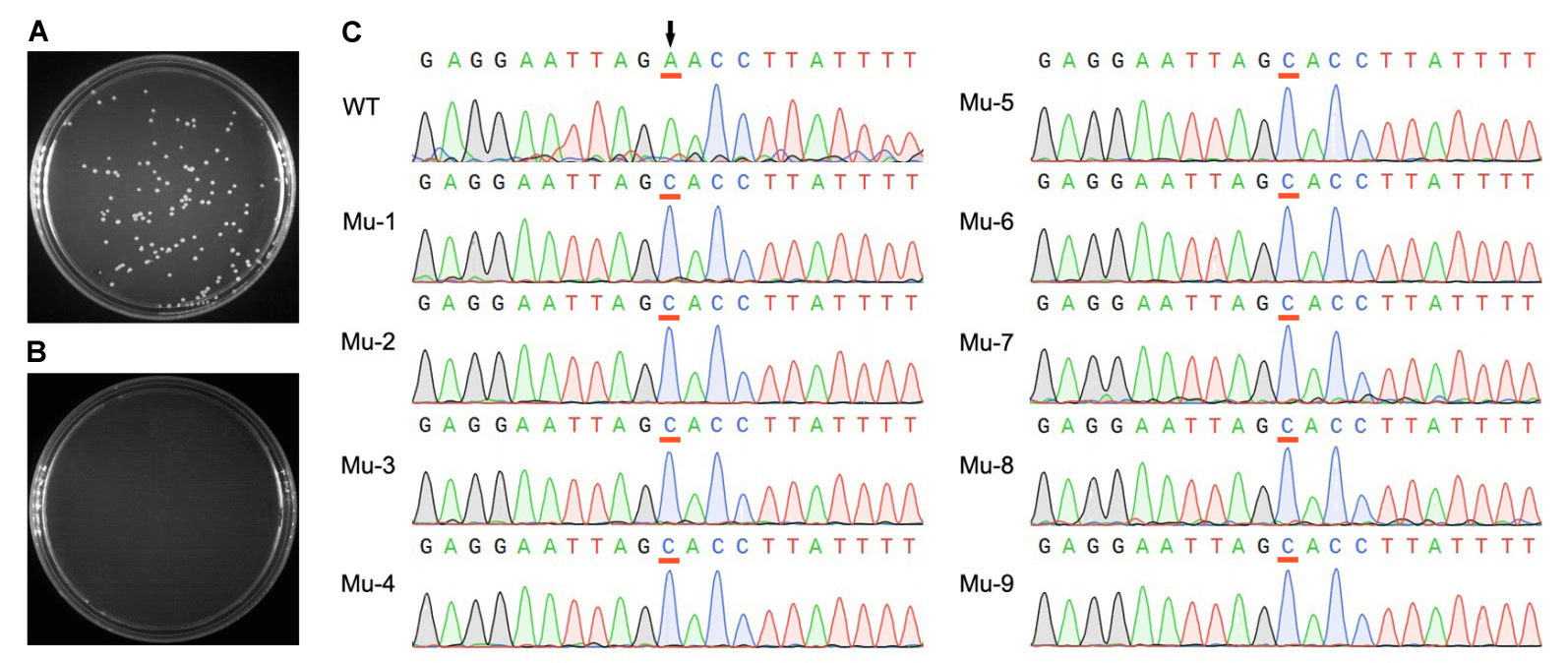
使用本产品的文献:
1. Wei JY, Li WM, Zhou LL, Lu QN, He W.
J Pineal Res
2015 May;58(4):429-38.
(IF 14.528)
2. Niu K, Fang H, Chen Z, Zhu Y, Tan Q, Wei D, Li Y, Balajee AS, Zhao Y
Autophagy.
16(4):724-734.
(IF 9.77)
3. Zhou ZJ, Dai Z, Zhou SL, Hu ZQ, Chen Q, Zhao YM, Shi YH, Gao Q, Wu WZ, Qiu SJ, Zhou J, Fan J.
Cancer Res
2014 May 15;74(10):2750-62.
(IF 9.727)
4. Yu Liang, Chun-Dong Zhang, Cheng Zhang, Dong-Qiu Dai
Gastric Cancer.
doi: 10.1007/s10120-019-01002-1.
(IF 7.088)
5. Rong ZH, Chang NB, Yao QP, Li T, Zhu XL, Cao Y, Jiang MJ, Cheng YS, Jiang R, Jiang J
MOL THER-NUCL ACIDS.
18:999-1008.
(IF 7.032)
6. Yong Lan, Min Dong, Yongjun Li, Yongpeng Diao, Zuoguang Chen, Yangfang Li
MOL THER-NUCL ACIDS.
doi: 10.1016/j.omtn.2021.01.023.
(IF 7.032)
7. Yang Y, Wu N, Tian S, Li F, Hu H, Chen P, Cai X, Xu L, Zhang J, Chen Z, Ge J, Yu K, Zhuang J.
Cell Death Dis
2016 Nov 17;7(11):e2473.
(IF 6.304)
8. Zhang YX, Yan YF, Liu YM, Li YJ, Zhang HH, Pang M, Hu JX, Zhao W, Xie N, Zhou L, Wang PY, Xie SY.
Cell Death Dis
2016 Dec 22;7(12):e2528.
(IF 6.304)
9. Huang Z, Li Q, Luo K, Zhang Q, Geng J, Zhou X, Xu Y, Qian M, Zhang JA, Ji L, Wu J
Cell Death Dis.
10(5):372.
(IF 6.304)
10. Yufeng Hu, Yangping Li, Jianfeng Weng, Hanmei Liu, Guowu Yu, Yinghong Liu, Qianlin Xiao, Huanhuan Huang, Yongbin Wang, Bin Wei, Yao Cao, Ying Xie, Tiandan Long, Hui Li, Junjie Zhang, Xinhai Li, Yubi Huang
Plant J.
doi: 10.1111/tpj.15043.
(IF 6.141)
11. Zhao F, Wang M, Li S, Bai X, Bi H, Liu Y, Ao X, Jia Z, Wu H.
Oncogenesis
2015 Mar 16;4:e143.
(IF 6.119)
12. Huang L, Hu X, Zhou M, Yang Y, Qiao J, Wang D, Yu J, Cui Z, Zhang Z, Zhang XE, Wei H.
J Clin Microbiol
2014 Jun;52(6):1947-53.
(IF 5.897)
13. Zhao H, Li H, Du W, Zhang D, Ge J, Xue F, Zhou Z, Yang R.
BRIT J HAEMATOL
2014 Sep;166(5):767-73.
(IF 5.518)
14. Ying-Xian Goh, Peifei Li, Meng Wang, Marko Djordjevic, Cui Tai, Hui Wang, Zixin Deng, Zhaoyan Chen, Hong-Yu Ou
Microbiol Spectr.
doi: 10.1128/spectrum.00320-22.
(IF 5.465)
15. Yuhang Sun, Guiying Zhai, Rui Li, Weinan Zhou, Yumao Li, Zhiping Cao, Ning Wang, Hui Li, Yuxiang Wang
Front Cell Dev Biol.
doi: 10.3389/fcell.2020.00349.
(IF 5.201)
16. Yang L, Shi LP, Chen HJ, Tong XK, Wang GF, Zhang YM, Wang WL, Feng CL, He PL, Zhu FH, Hao YH, Wang BJ, Yang DL, Tang W, Nan FJ, Zuo JP.
Acta Pharmacol Sin
2014 Mar;35(3):410-8.
(IF 5.064)
17. Song ZB, Bao YL, Zhang Y, Mi XG, Wu P, Wu Y, Yu CL, Sun Y, Zheng LH, Huang YX, Liu B, Li YX.
Biochem J
2011 Jun 1;436(2):457-67.
(IF 4.097)
18. Li YY, Bao YL, Song ZB, Sun LG, Wu P, Zhang Y, Fan C, Huang YX, Wu Y, Yu CL, Sun Y, Zheng LH, Wang GN, Li YX.
PLoS One
2012;7(5):e35030.
(IF 2.74)
19. Guo CJ, Yang LS, Zhang YF, Wu YY, Weng SP, Yu XQ, He JG.
PLoS One
2012;7(7):e41092.
(IF 2.74)
20. Fei X, Qi M, Wu B, Song Y, Wang Y, Li T.
FEBS Lett
2012 Feb 17;586(4):392-7.
(IF 3.057)
21. Qin Y, Fang Z, Pan F, Zhao Y, Li H, Wu H, Meng X.
Biotechnol Lett
2012 May;34(5):895-9.
(IF 1.977)
22. Li F, Jiang Z, Wang K, Guo J, Hu G, Sun L, Wang T, Tang X, He L, Yao J, Wen D, Qin X, Zhang L.
Gene
2012 Jul 25;503(2):200-7.
(IF 2.984)
23. Liu Y, Zhang C, Chen J, Guo L, Li X, Li W, Yu Z, Deng J, Zhang P, Zhang K, Zhang L.
PLANT PHYSIOL BIOCH
2013 Mar;64:92-8.
(IF 3.72)
24. Li X, Lu Y, Chen Y, Lu W, Xie X.
BMC Cancer
2013 Apr 30;13:216.
(IF 3.15)
25. Wang G, Sai K, Gong F, Yang Q, Chen F, Lin J.
Mol Med Rep
2014 May;9(5):1799-805.
(IF 2.1)
26. Peng Y, Guo JJ, Liu YM, Wu XL
BIOSCIENCE REP
2014 Jun 25;34(3).pii: e00112.
(IF 2.942)
27. Guan Y, Guo L, Yang E, Liao Y, Liu L, Che Y, Zhang Y, Wang L, Wang J, Li Q.
Virology
2014 Sep;464-465:1-10.
(IF 2.819)
28. Zhang XW, Zhang BY, Wang SW, Gong DJ, Han L, Xu ZY, Liu XH.
J THORAC CARDIOV SUR
2014 Oct;148(4):1700-1708.
(IF 4.451)
29. Liu Y, Zhou J, Pan JA, Mabiala P, Guo D.
Mol Biotechnol
2014 Oct;56(10):890-902.
(IF 2.022)
30. Jiang W, Min J, Sui X, Qian Y, Liu Y, Liu Z, Zhou H, Li X, Gong Y.
LEUKEMIA LYMPHOMA
2015 Feb;56(2):460-71.
(IF 2.969)
31. Guo X, Qi Y, Huang Y, Liu Z, Ma Y, Shao Y, Jiang S, Sun Z, Ruan Q.
FEBS Lett
2015 Feb 13;589(4):440-6.
(IF 3.057)
32. Yang S, Fan R, Shi Z, Ji K, Zhang J, Wang H, Herrid M, Zhang Q, Yao J, Smith GW, Dong C.
J Anim Sci
2015 Apr;93(4):1622-31.
(IF 2.092)
33. Wei JY, Lu QN, Li WM, He W.
Brain Res
2015 Apr 16;1604:15-24.
(IF 2.733)
34. Zhao Y, Wang P, Meng J, Ji Y, Xu D, Chen T, Fan R, Yu X, Yao J, Dong C.
Int J Mol Sci
2015 May 14;16(5):10921-33.
(IF 4.556)
35. Li X, Pan Q, Wan X, Mao Y, Lu W, Xie X, Cheng X.
BMC Cancer
2015 Jul 8;15:509.
(IF 3.15)
36. Wang XL, Zhang T, Wang J, Zhang DB, Zhao F, Lin XW, Wang Z, Shi P, Pang XN.
PLoS One
2015 Aug 27;10(8):e0136049.
(IF 2.74)
37. Jiang S, Qi Y, He R, Huang Y, Liu Z, Ma Y, Guo X, Shao Y, Sun Z, Ruan Q.
Gene
2015 Oct 1;570(1):108-14.
(IF 2.984)
38. Guo X, Huang Y, Qi Y, Liu Z, Ma Y, Shao Y, Jiang S, Sun Z, Ruan Q.
Arch Virol
2015 Oct;160(10):2483-90.
(IF 2.243)
39. Xiao Y, Yao Y, Jiang H, Lu C, Zeng S, Yu L.
Biopharm Drug Dispos
2015 Nov;36(8):520–8.
(IF 1.663)
40. Li C, Wang Y, Lu S, Zhang Z, Meng H, Liang L, Zhang Y, Song B.
Mol Med Rep
2015 Nov;12(5):7072-8.
(IF 2.1)
41. Zhou Y, Wang M, Wu J, Jie Z, Chang S, Shuang T.
J Ovarian Res
2015 Dec;8:23.
(IF 2.989)
42. Cheng S, Liang X, Wang Y, Jiang Z, Liu Y, Hou W, Li S, Zhang J, Wang Z.
EXP BIOL MED
2016 Jan;241(2):205-15.
(IF 3.139)
43. Yang D, Yan R, Zhang X, Zhu Z, Wang C, Liang C, Zhang X.
Am J Transl Res
2016 Mar 15;8(3):1551-9.
(IF 3.375)
44. Mi S, Qin XW, Lin YF, He J, Chen NN, Liu C, Weng SP, He JG, Guo CJ.
SCI REP-UK
2016 May 26;6:26581.
(IF 3.998)
45. Li Z, Yin H, Hao S, Wang L, Gao J, Tan X, Yang Z.
SCI REP-UK
2016 Jun 2;6:27105.
(IF 3.998)
46. Shao Y, Qi Y, Huang Y, Liu Z, Ma Y, Guo X, Jiang S, Sun Z, Ruan Q.
J BIOSCI BIOENG
2016 Jun;41(2):183-92.
(IF 2.366)
47. Xu Y, Liu F, Liu J, Wang D, Yan Y, Ji S, Zan J, Zhou J.
SCI REP-UK
2016 Jun 2;6:27123.
(IF 3.998)
48. Gao Z, Zhang H, Hu F, Yang L, Yang X, Zhu Y, Sy MS, Li C.
Cell Signal
2016 Jun;28(6):652-62.
(IF 3.968)
49. Chen Q, Zhang J, Zhang F, Guo H, Fang Q.
Virol Sin
2016 Aug;31(4):314-23.
(IF 3.242)
50. Fang T, Wu Q, Zhou L, Mu S, Fu Q.
Exp Cell Res
2016 Sep 10;347(1):74-82.
(IF 3.383)
51. Huang Y, Li Y, Wang FF, Lv W, Xie X, Cheng X.
PLoS One
2016 Sep 14;11(9):e0162378.
(IF 2.74)
52. Zhang J, Liu Y, Zhu Z, Yang S, Ji K, Hu S, Liu X, Yao J, Fan R, Dong C.
Animal
2017 Feb;11(2):236-243.
(IF 2.4)
53. Zhang Y, Yao L, Xu X, Han H, Li P, Zou D, Li X, Zheng L, Cheng L, Shen Y, Wang X, Wu X, Xu J, Song B, Xu S, Zhang H, Cao H
Virus Res
2018 Aug 15;255:55-67.
(IF 2.934)
54. Jiang YL, Zhao ZY, Li BR, Yang F, Li J, Jin XW, Wang H, Yu ED, Sun SH, Ning SB
BMC Med Genet
2018 Aug 9;19(1):141.
(IF 1.585)
55. Li P, Huang Z, Wang J, Chen W, Huang J
ONCOTARGETS THER.
12:1603-1611.
(IF 3.337)
56. Li W, Fang J, Shen J, Liu X, Hou J, Zhu Y, Ma X
BIOCHEM BIOPH RES CO.
509(2):603-610.
(IF 2.985)
57. Li X, Xiao J, Fan Y, Yang K, Li K, Wang X, Lu Y, Zhou Y
J Endocrinol.
242(3):185-197.
(IF 4.041)
58. Zhu XX, Li JH, Cai JP, Hou X, Huang CS, Huang XT, Wang JQ, Li SJ, Xu QC, Yin XY
Cancer Sci.
110(10):3110-3121.
(IF 4.966)
59. Tian N, Shangguan W, Zhou Z, Yao Y, Fan C, Cai L
J Cancer.
10(24):6074-6087.
(IF 3.565)
60. Zong H, Zhou H, Xiang Y, Wu G
Oncol Lett.
18(6):6822-6828.
(IF 2.311)
61. Zhu B, Sun X, Nie X, Liang P, Gao X
INSECT BIOCHEM MOLEC.
117:103283.
(IF 3.827)
62. Bin Zhu, Xi Sun, Ximan Nie, Pei Liang, Xiwu Gao
INSECT BIOCHEM MOLEC.
doi: 10.1016/j.ibmb.2019.103283.
(IF 3.827)
63. Min Li, Ling-Yan Jiang, Qin Liu, Yuan-Hang Wu, Guo-Dao Liu, Yin-Hua Chen, Li-Juan Luo
Biotechniques.
doi: 10.2144/btn-2019-0141.
(IF 1.541)
64. Zhi-Fei Liu, Jian-Fei Ji, Xiao-Feng Jiang, Tong Shao, Dong-Dong Fan, Xin-Hang Jiang, Ai-Fu Lin, Li-Xin Xiang, Jian-Zhong Shao
FASEB J.
doi: 10.1096/fj.201902833R.
(IF 4.966)
65. Xiuli Gao, Hongguang Bao, Likun Liu, Wenbin Zhu, Liping Zhang, Liling Yue
Oncol Rep.
doi: 10.3892/or.2020.7554.
(IF 3.417)
66. Shuping Zhang, Yan Wang, Ping Wang, Jin Xuan
MOL CELL PROBE.
doi: 10.1016/j.mcp.2020.101653.
(IF 1.951)
67. Yanan Wang, Houshuang Zhang, Li Luo, Yongzhi Zhou, Jie Cao, Xuenan Xuan, Hiroshi Suzuki, Jinlin Zhou
PLOS NEGLECT TROP D .
doi: 10.1371/journal.pntd.0009074.
(IF 4.367)
68. Hanqun Liu, Yuxuan Yong, Xingjian Li, Panghai Ye, Kai Tao, Guoyou Peng, Mingshu Mo, Wenyuan Guo, Xiang Chen, Yangfu Luo, Yuwan Lin, Jiewen Qiu, Zhiling Zhang, Liuyan Ding, Miaomiao Zhou, Xinling Yang, Lin Lu, Qian Yang, Pingyi Xu
Neurosci Bull.
doi: 10.1007/s12264-022-00818-9.
(IF 4.326)
69. Zhengyun Hu, Guoping Zhou
Comput Math Methods Med.
doi: 10.1155/2022/9588740.
70. Linxia Sun, Xinghua Rong, Xing Liu, Zhenpeng Yu, Qian Zhang, Wenhua Ren, Guang Yang, Shixia Xu
BMC Genomics.
doi: 10.1186/s12864-022-09024-3.
(IF 3.594)
71. Xihui Chen, Fangfang Liu, Kun Chen, Yufeng Wang, Anan Yin, Xiaowei Kang, Shanming Yang, Hanwen Zhao, Songqi Dong, Yunqing Li, Jing Chen, Yuanming Wu
CNS Neurosci Ther.
doi: 10.1111/cns.13943.
(IF 4.074)